식품의약품안전처(이하 식약처)가 국산 복제약(제네릭 의약품)의 심사 단계에서 불순물을 파악하는 등 품질과 안전 관리 수준을 대폭 강화하기로 했다.
식약처는 16일 제네릭 의약품 국제경쟁력 강화를 위한 민관협의체 논의 결과 이런 내용을 포함한 21개 과제를 도출했다고 밝혔다. 식약처는 우선 국내 유통 중인 복제약의 품질을 확보하고자 전 공정을 위탁해 제조할 때에도 의약품 제조 및 관리 기준(GMP) 적격성 여부를 확인하기로 했다.
현행 규정에서는 실제 제조하는 수탁자 품목만 GMP를 제출하게 했으나 앞으로는 위탁자 품목도 GMP 자료를 제출해야 한다. 그간 업체 자율로 관리하던 허가 복제약의 제조 방법 변경은 품질이나 약효에 미치는 영향에 따라 사전 변경 허가하는 식으로 안전관리를 강화하기로 했다.
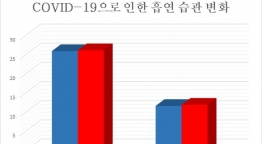
특히 국내에서 잇따라 문제가 됐던 의약품 불순물 문제도 재발하지 않도록 할 방침이다. 국내에서는 고혈압 치료제, 위장약, 당뇨병 치료제 등에서 발암 추정물질인 N-니트로소디메틸아민(NDMA)가 기준치 이상 검출돼 논란에 휩싸인 바 있다.
이에 식약처는 완제의약품 심사단계에서 원료의약품의 NDMA 등 불순물과 같은 위험요인을 사전에 파악해 품질의 사각지대를 해소하기로 했다. 완제의약품 심사 시 원료의약품에 관한 품질심사를 병행함으로써 허가심사의 실효성을 높일 방침이다.
복제약도 쉽게 파악할 수 있도록 관련 정보도 투명하게 공개하기로 했다. 동일 제조소에서 제조되고, 동일 동등성시험 자료로 허가된 제네릭의약품의 목록을 묶음정보로 만들어 식약처 홈페이지, 모바일웹에 공개할 예정이다.
Copyright © 의약일보